


INTRODUCTION
Good's syndrome, a combination of thymoma and adult-onset immunodeficiency, is a rare cause of combined B- and T-cell immunodeficiency. It is classified as a primary immunodeficiency1), and patients usually present with recurrent infections or a mediastinal mass2). The precise cause and pathogenesis of this disorder are unclear, although there is evidence that the primary defect is associated with bone marrow defects, including pre-B cell arrest and impaired maturation of erythroid and myeloid precursors3).
Patients with Good's syndrome are susceptible to infections with encapsulated bacteria and opportunistic viruses and fungi. We describe a patient with Good's syndrome who developed a severe infection subsequent to a thymectomy to remove a thymoma. Our patient had granulomatous lung disease, which became infected with Pneumocystis jirovecii.
CASE REPORT
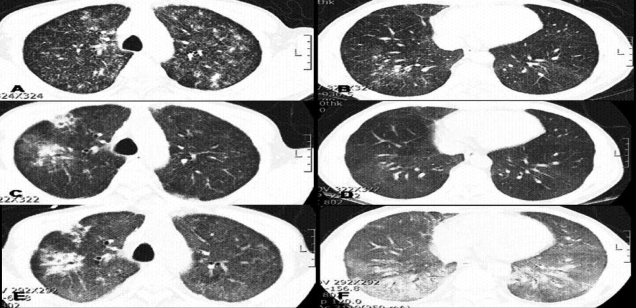
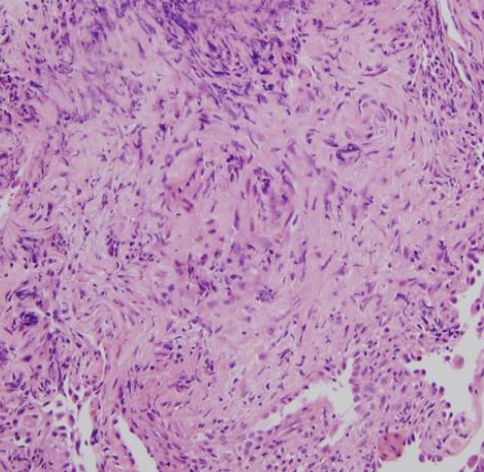
A 53-year-old man presented at our hospital with a 7-day history of exertional dyspnea. He also complained of a productive cough, which he had had for about 40 days. He had undergone an extended thymectomy to remove a lymphoepithelioid type of thymoma 5 months previously and had received 45 Grays of adjuvant radiotherapy. Two months before admission, he was diagnosed with sarcoidosis based on the results of an imaging study (Figure 1) and a transbronchial lung biopsy (Figure 2). However, his medication was changed to an anti-tuberculosis chemotherapeutic regimen 10 days before admission because a new cavitary lesion was detected on high-resolution computed tomography (HRCT).
On admission to our hospital, he had a blood pressure of 90/60 mmHg, a body temperature of 39.3Ōäā, a pulse rate of 152 beats per minute, and a respiration rate of 32 breaths per minute. Physical examination revealed inspiratory crackles in both lower lung fields. A chest roentgenogram showed haziness and disseminated nodules in both lung fields and conglomerated cavities in the right upper lobe.
Computerized tomography (CT) of the chest revealed multiple micronodular densities and multiple cavitary nodules in the right upper lobe. Comparison with CT obtained 10 days previously showed that his condition had worsened. In addition, new regions of ground glass opacity (GGO) were detected in both lower lobes (Figure 1).
Laboratory analyses revealed a white blood cell count of 3500 cells/mm3 (79.6% segmental forms), a hemoglobin of 15.2 g/dL, a platelet cell count of 253,000 cells/mm3, an erythrocyte sedimentation rate of 36 mm/h, and a C-reactive protein level of 17.29 mg/dL. His hepatic function was normal, except for a mildly elevated aspartate aminotransferase (AST) level of 81 U/L. Renal function was normal and an HIV test was negative. Arterial blood gas analysis (fraction of inspired oxygen 0.44) revealed a pH of 7.44, a PaCO2 of 34.0 mmHg, a PaO2 of 60.5 mmHg, a bicarbonate (HCO3) level of 22.8 mmol/L, and an SaO2 of 91.9%. The patient was admitted to the intensive care unit.
We performed bronchoalveolar lavage (BAL) of the posterior basal segment of the right lower lobe to identify the causative organism. The cell count from the BAL fluid was 119/┬ĄL, and the cell differential showed 30% macrophages, 60% lymphocytes (CD4/CD8=7/46), and 10% neutrophils. Polymerase chain reaction (PCR) and direct florescent antibody (DFA) analyses of the lavage fluid were positive for Pneumocystis jirovecii. Culture for cytomegalovirus (CMV) was negative. Trimethoprim/sulfamethoxazole and methylprednisolone were administered because of the presence of P. jirovecii.
The result of a neutrophil dihydrorhodamine test, which assesses the immune function of polymorphonuclear cells from the oxidase peak, was normal.
The patient's cell counts were as follows: T lymphocytes (CD3), 290/┬ĄL; B lymphocytes (CD19), 0/┬ĄL; CD4, 148/┬ĄL, and CD8, 132/┬ĄL. His immunoglobulin levels were low: IgG, 507 mg/dL; IgA, 31 mg/dL; and IgM <3 mg/dL.
Although we performed a video-assisted thoracoscopic biopsy, the biopsy specimens revealed only diffuse thickening of the alveolar wall with fibrosis and lymphocyte infiltration. The analysis of a smear of the specimen for acid-fast bacilli and the immunohistochemical analysis for CMV, herpes simplex virus (HSV), and adenovirus antigens were negative. Having diagnosed Good's syndrome, we continued supportive ventilatory care and antibiotic treatment for acute respiratory distress syndrome.
Since the hypogammaglobulinemia persisted despite antibiotic treatment, the patient was given regular intravenous immunoglobulin (IVIG) infusions. However, the pneumonia and diarrhea did not improve and the infection worsened. Culture of the pleural fluid and material from the catheter tip showed the presence of Candida albicans. The patient died of septic shock and multi-organ failure caused by uncontrolled fungal infection during the third month after admission.
DISCUSSION
The patient had hypogammaglobulinemia and had previously undergone a thymectomy. He died of uncontrolled fungal infection that originated from damaged skin and a central venous catheter.
Good's syndrome (immunodeficiency with concomitant thymoma) is a rare cause of combined B- and T-cell immunodeficiency in adults. In 1954, Good first reported a thymoma coexistent with hypogammaglobulinemia2).
Patients with Good's syndrome usually present in the 4th or 5th decade of life, and the frequency in males and females is similar. The immunodeficiency may precede or follow the diagnosis of a thymoma2), but the immunologic abnormalities cannot be corrected by corticosteroid treatment or thymectomy4).
The combination of common variable immunodeficiency (CVID) and thymoma is known as Good's syndrome2). As this condition satisfies the criteria for a combined immunodeficiency presenting in infancy with severe lethal infections1), it may be regarded as a subset of CVID5). Diffuse interstitial lung disorders (granulomatous lung disease, lymphocytic interstitial pneumonia, follicular bronchiolitis, and bronchiolitis obliterans with organizing pneumonia) that are refractory to IVIG treatment occur in approximately 25% of patients with CVID. As these disorders are consistent with the diagnostic criteria for Good's syndrome6, 7), there is considerable debate regarding the nature and clinical course of granulomatous lung disease in patients such as ours8, 9).
Cases of non-necrotizing granulomatous lung disease that are resistant to corticosteroid therapy have been reported. Bates et al.10) proposed that granulomatous lymphocytic lung disease shortens the survival of CVID patients.
The cause of granulomatous disease in CVID patients is unclear. Some believe it is a sarcoidosis of immunocompromised patients, whereas others contend that it is a unique clinical disorder11).
Good's syndrome is classified as a distinct primary immunodeficiency1). Although the cause and pathogenesis of this disorder are unknown, its main characteristics are hypogammaglobulinemia; a low B-cell count or the absence of B cells; variable defects in cell-mediated immunity such as CD4+ T-cell lymphopenia and a reduced T-cell proliferative response to mitogens; and reduced serum levels of IgG, IgA, and IgM4). A reduced mature B-cell count in bone marrow has also been reported12).
Since cell-mediated immunity is compromised, individuals with Good's syndrome are susceptible to bacterial infections, particularly infections with encapsulated organisms, and opportunistic viral and fungal infections. Thymoma occurs in 10% of patients with adult-onset hypogammaglobulinemia, whereas 3~6% of thymoma patients have hypogammaglobulinemia 4).
Tarr et al.2) reported that sinopulmonary infection with Haemophilus influenza was the most common infectious disease among 51 patients with immunodeficiency and thymoma. Other infectious diseases present in their patients were CMV, Pneumocystis jirovecii, infectious diarrhea, and tuberculosis. Although systemic fungal infections are not characteristic of Good's syndrome, mucocutaneous candidiasis occurs in 24% of cases4). Autoimmune diseases such as myasthenia gravis, pure red-cell aplasia, pernicious anemia, diabetes mellitus, and idiopathic thrombocytopenia also occur1). Although our patient did not have an autoimmune disease, he had a history of frequent respiratory infections that were aggravated after the thymectomy. The patient died of Candida infection.
The treatment of patients with Good's syndrome includes resection of the thymoma and immunoglobulin replacement to maintain adequate IgG levels. IVIG improves the control of infections, reduces the duration of hospitalization, decreases the quantity of antibiotics used, and might prevent chronic lung damage and bronchiectasis2, 13, 14).
According to a follow-up report2), 29 of 50 Good's syndrome patients died. A favorable response during the follow-up period was observed for 23 of 30 patients, although the response was often transient. Nine of the 22 responders and six of the seven non-responders died. Hermaszewski et al. reported that the 5- and 10-year survival rates were 70 and 33%, respectively15).
In summary, we report a rare case of granulomatous lung disease similar to that observed in CVID, of which Good's syndrome is a subset. Good's syndrome should be suspected in patients with thymoma or CVID who develop repeated respiratory infections, especially opportunistic infections.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





