


INTRODUCTION
Idiopathic adulthood ductopenia (IAD) was first proposed as a condition characterized by chronic cholestasis by Ludwig et al. in 1988 [1]. Currently, the diagnosis of IAD is considered with adult or adolescent onset, biochemical evidence of cholestatic liver disease, evidence of ductopenia by liver biopsy with normal cholangiography, and an unknown etiology. A diagnosis of IAD requires exclusion of other conditions with chronic cholestasis that are associated with ductopenia [1-3]. To date, less than 100 cases have been reported in the medical literature, with only 1 prior case in Korea [4]. We report a case of idiopathic adulthood ductopenia with a brief review of the medical literature.
CASE REPORT
A 19-year-old man who had abnormal liver function tests (LFTs) was referred to the outpatient clinic. Four years and 1 month earlier, abnormal LFTs were detected repeatedly, but the patient remained asymptomatic. He had no history of neonatal or infantile liver disease, previous drug treatments, jaundice, itching, or abdominal pain. There was no history of smoking, alcohol drinking and any medication treatments. His mother was diagnosed with chronic type B hepatitis. Physical examination showed no abnormal findings. Laboratory tests revealed the following findings: hemoglobin 14.5 g/dL, white blood count 7090/mm3, platelets 376,000/mm3, alanine aminotransferase (ALT) 147 IU/L, aspartate aminotransferase (AST) 123 IU/L, alkaline phosphatase (ALP) 1345 IU/L, ╬│-glutamyl transpeptidase (GGT) 576 IU/L, total bilirubin 0.9 mg/dL and albumin 4.0 g/dL. Serum iron, transferrin saturation, ferritin, alpha 1-antitrypsin, and ceruloplasmin were normal. Serological tests for anti-smooth muscle, anti-neutrophil-cytoplasm, anti-nuclear, anti-mitochondrial, anti-liver-kidney microsome antibodies, hepatitis B surface antigen, and hepatitis C virus serology were negative.
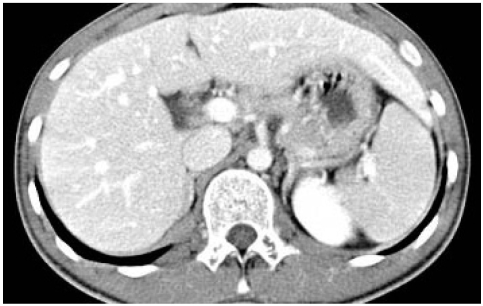
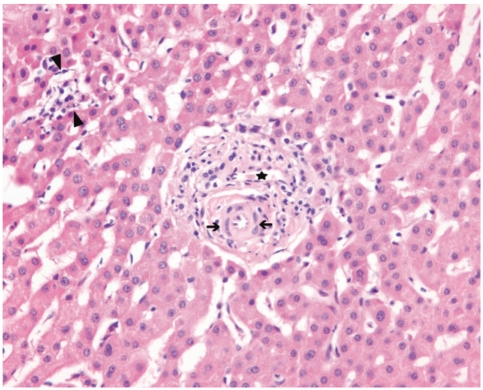
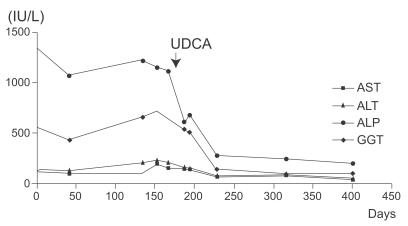
An abdominal CT showed normal echogenicity of the liver. No evidence of cholelithiasis or biliary dilation was noted (Fig. 1). During a six-month follow-up period, his LFTs showed persistent elevation of the transaminases and alkaline phosphatase without clinical symptoms. He was admitted to the hospital for further evaluation. An endoscopic retrograde cholangiopancreatography (ERCP) demonstrated completely normal extrahepatic bile ducts and minimal irregularities of the intrahepatic bile ducts in the left lobe (Fig. 2). A colonoscopy was normal. Percutaneous liver biopsy showed a lack of interlobular bile ducts. In 19 portal tracts, only eight bile ducts were found. Mild chronic inflammatory infiltrates consisting primarily of lymphocytes and liver parenchymal spotty necrosis were also found (Fig. 3). The patient was diagnosed with idiopathic adulthood ductopenia. Treatment was started with ursodeoxycholic acid at doses of 300 mg twice daily and changed to 200 mg twice daily with vitamins. Eight months later, the laboratory test results were AST, 52 IU/L; ALT, 64 IU/L; GGT, 112 IU/L; ALP, 208 IU/L; and total bilirubin 0.9 mg/dL (Fig. 4).
DISCUSSION
Primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC) are the most common causes of chronic progressive cholestasis in adults [5,6]. However, a small number of patients who present with chronic cholestasis, lack anti-mitochondrial antibodies for PBC and have cholangiographic abnormalities consistent with PSC. These groups of patients often are thought to have AMA-negative PBC, small-duct PSC, or idiopathic adulthood ductopenia [7]. Ludwig reported the causes of small duct biliary diseases in 2,082 cases diagnosed between 1988 and 1994 at the Mayo Clinic. While PBC and PSC accounted for more than 90% of the cases with small duct biliary diseases, IAD was present in only 1% of cases [2]. In Korea, only one case in 2004, a 28-year-old woman who presented with pruritus and jaundice, has been reported [4].
The etiology of IAD is unknown. Several possible causes have been suggested, including late onset nonsyndromic paucity of the intrahepatic bile ducts, small-duct PSC without large duct involvement and without evidence of inflammatory bowel disease, nonsuppurative viral cholangitis, hepatitis C virus, and autoimmune cholangitis or cholangitis in autoimmune hepatitis, in the absence of typical autoantibodies [1,2,8,9]. Typically, IAD is more common in males (M:F, 1.8:1), and it is usually diagnosed in young or middle-aged adults (15-77 years, median age: 34 years, 30 years for male, 36 years for female) [1,2]. However, each etiologic group appears to have a different age distribution. Late onset nonsyndromic paucity of intrahepatic bile ducts usually is diagnosed in the teenage years and nonsuppurative viral cholangitis in adults more than 40 years of age [1,2]. This case appears to belong to the late-onset nonsyndromic paucity of intrahepatic bile ducts, as abnormal LFTs have been identified in teenagers.
Ductopenia by liver biopsy is the criterion necessary for the diagnosis of IAD. Ductopenia is defined as the loss of interlobular and septal bile ducts of more than 50% among 20 or more portal tracts [1,2,10]. Clinical symptoms include jaundice, pruritus and fatigue. Approximately one-third of patients present with episodic jaundice and pruritus, which gradually worsens. In other patients, cholestasis progresses incessantly, although the rate of deterioration varies [7]. Laboratory findings demonstrate the cholestatic profiles. Direct bilirubin levels are frequently increased. ALP and GGT are increased more than three times the upper limit of normal. Transaminase levels vary between normal and a 10-fold increases [1,2,7]. In this case, there was a persistent cholestatic pattern of LFTs with a normal bilirubin.
Treatments for IAD depend on the etiology and the severity of the clinical course [11,12]. IAD related to autoimmune hepatitis or viral hepatitis requires specific treatments. In advanced cases, liver transplantation is the treatment of choice [13,14]. In cases that are not severe, ursodeoxycholic acid (UDCA) can be used for treatment. A beneficial effect with the treatment of UDCA has been suggested, but its impact on the progression of the disease is unknown [11,12].
The clinical outcome may be dependent on the cause of the disease and on the degree of ductopenia. Most patients with a late-onset nonsyndromic paucity of the intrahepatic bile ducts and small-duct primary sclerosing cholangitis may have progressive disease, but this may be self-limited in patients with virus-related cholangitis and autoimmune cholangitis [2,11,12]. Typically, IAD eventually progresses to end-stage liver disease. However, some patients with mild to moderate loss of intrahepatic bile ducts, less than 50% based on a liver biopsy, have a benign course [2,8,11]. Mild IAD, also termed mild idiopathic biliary ductopenia, does not meet the current diagnostic criteria for IAD as no cases have had ductopenia involving 50% of the portal tracts [11]. However, they appear to have features consistent with this diagnosis. Our patient had only abnormal LFTs without any symptoms, but had a ductopenia of more than 50%. Whether his course will be benign is not clear at this time. IAD is a progressive disease and our patient may have been diagnosed at an early stage.
In conclusion, little is known about IAD. The pathogenesis, causes, clinical course, and best treatments remain unclear. IAD is not a specific single entity but a syndrome associated with variable etiologies and variable clinical outcomes.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





