


INTRODUCTION
Socioeconomic burden of rare genetic diseases is increasing. There have been numerous attempts to treat genetic diseases with various methods. However, they were not overly successful till now. Recently, the technology of clustered regularly interspaced short palindromic repeats (CRISPR) emerges as a promising tool to correct genetic abnormalities. This technique is being heralded for precision and accuracy in genetic editing. In this review, we recapitulate the history and recent progress made in the area of CRISPR technology. In the first part of the review, we summarize the history and action mechanism of CRISPR. In the second part of the review, we deliberate upon assorted clinical applications of CRISPR, from the standpoint of recent feasibility and future possibilities. In the third part, we discuss about future perspective of CRISPR technology. Ideal combination of CRISPR technology and induced pluripotent stem cell (iPSC) may bring new CRISPR-based clinical applications into clinics in near future.
PART 1. HISTORY AND MECHANISMS OF ACTION
In light of the heterogeneity of disease manifestations among patients, the field of precision medicine has attracted a great deal of interest, especially following a new initiative launched in 2015. The Precision Medicine Initiative (PMI), which involves investment in medical research in the United States on a national scale, envisions treatment and prevention of diseases on an individual basis, according to differences in genes and environmental and lifestyle factors [1,2]. The short-term goals of the PMI include combating cancers, while the accrual of knowledge pertaining to health and diseases represents the long-term focus. The aggregation of personalized information pertaining to genomics, proteomics, and phenotypical parameters is expected not only to provide a better understanding of health and disease, but also to change our approach to risk assessment, diagnostic tests, and therapeutic interventions [3]. However, despite major progress in gene sequencing and profiling, based on advances in technologies such as high-throughput sequencing, precise gene editing remains challenging; this difficulty has hindered the translation of information into clinical applications. As a consequence, the demand for targeted, straightforward, and affordable genetic engineering tools continues to grow.
Over the past several decades, scientists have revolutionized genetic engineering techniques to allow modulation of their function. Since Watson and Crick elucidated the structure of the DNA double helix, many researchers have focused on changing the genome according to particular scientific needs. To achieve this, a platform that can identify the target sequence of interest, specifically cleave that region of the DNA, and alter the sequence at the cleavage site is required. Endogenous site-specific DNA-protein complexes and natural DNA repair pathways from multiple species have been exploited to create various gene engineering toolkits. Among these, the most rapidly evolving technology involves CRISPR and CRISPR-associated nuclease 9 (Cas9) (CRISPR-Cas9), which was selected as ScienceŌĆÖs Breakthrough of the Year in 2015.
CRISPR-Cas9 in the bacterial adaptive immune system
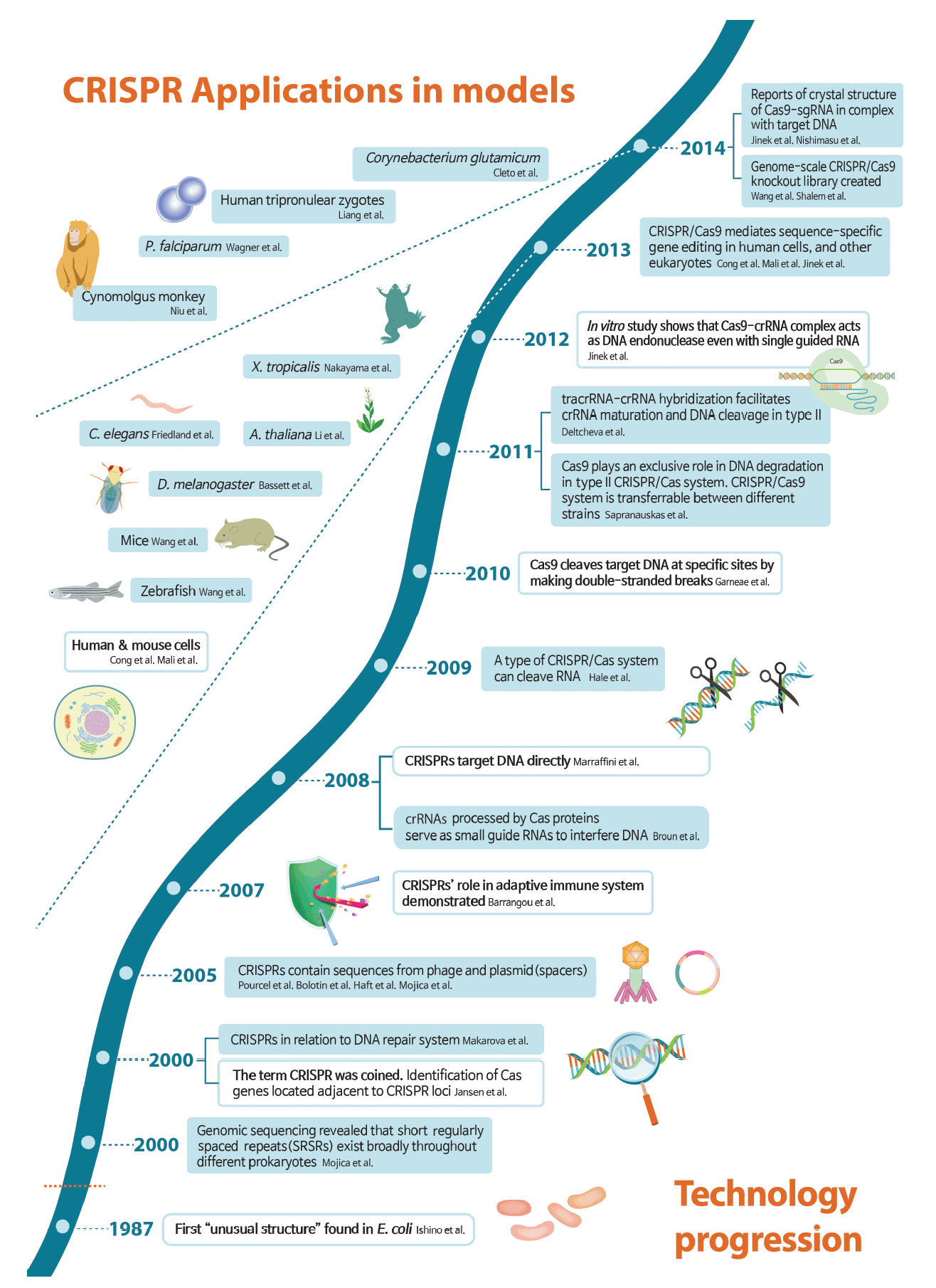
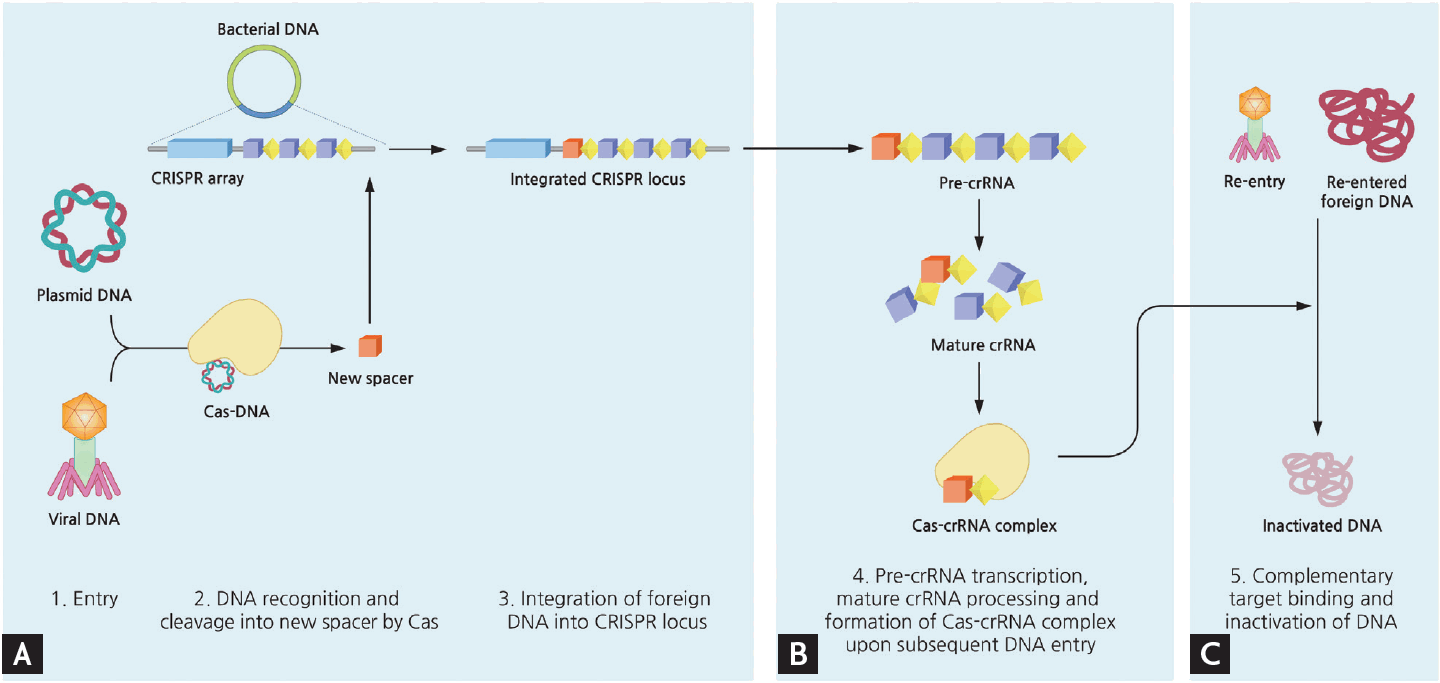
CRISPR is derived from the prokaryotic adaptive immune system (Fig. 1) [4-36]. The distinctive clustered repeats were originally recognized in Escherichia coli by Ishino et al. [5] in 1987, and were later found to include unique barcode-like sequences of viral or plasmid origin, termed spacers (Fig. 2) [6-8]. In 2007, the hypothesized role of the repeats in adaptive defense was confirmed by experimental demonstration of spacer integration into the bacterial genome following phage challenge, as well as alteration of sensitivity to subsequent phage infection dependent upon the spacer content [4]. Subsequent studies revealed that CRISPR works in sync with the Cas gene, in the vicinity of the CRISPR locus, to cleave DNA or RNA sequences [9,10] targeted by a small guide RNA [11]. Based on these findings, multiple studies sought to identify the components of the CRISPR-Cas system and apply this knowledge to sequence-specific gene engineering.
Mechanism underlying CRISPR-Cas9 gene editing
There are six putative CRISPR systems; the three main types (types I to III) were discovered first, with three additional types (types IV to VI) being identified more recently [37,38]. During the processes of immunity, adaptation, expression, and interference, each type acts according to distinct mechanisms to ensure DNA recognition and cleavage [39].
Type I uses a large complex of Cas proteins, encoded by the Cas3 gene, which show separate helicase and DNase activities. Similarly, type III uses repeat-associated mysterious proteins, which constitute a large superfamily of Cas proteins. Types I, III, and IV are categorized as class 1 based on their multi-subunit effector complexes. By contrast, class 2 systems (comprising types II, V, and VI) each have a single-subunit effector. Type II uses only a single protein (Cas9) for its nuclease activity; the same is true for types V and VI, but with Cas9-like proteins. Owing to their simplicity, class 2 systems have been adopted for genome engineering [40-42], and only the bacterial type II CRISPR-Cas9 system has been utilized for RNA-guided engineering nucleases [43,44].
As noted above, type II CRISPR-Cas9 systems use a single endonuclease, Cas9. This enzyme acts in concert with two guide RNAs: CRISPR RNA (crRNA) and trans-acting CRISPR RNA (tracrRNA) (Fig. 3) [45]. To simplify the system and improve its utility, scientists employed a linker loop to engineer a dual tracrRNA:crRNA (called single guide RNA [sgRNA]), which participates in sequence-specific DNA cleavage with Cas9 [12]. Short 2 to 5 bp conserved sequences known as proto-spacer adjacent motifs, located on the side opposite to that of the RNA-DNA hybridization, are required for Cas9-DNA recognition [6]. Once recognition occurs, double-stranded DNA cleavage is performed by two Cas9 domains: the HNH domain, which cleaves the strand complementary to the crRNA-guide sequence, and the RuvC-like domain, which cleaves the noncomplementary strand [12]. Via this mechanism, programmed nucleases with customized sgRNA can cleave genomic DNA at specific loci, enabling precise genome editing.
Cas9 protein can be easily re-targeted to new DNA sequences by changing a small portion of the sequence of the accompanying RNA guide, which base-pairs directly with target DNA [46]. Another potential advantage of Cas9 is its ability to introduce several double-strand breaks (DSBs) within the same genome (also referred to as multiplexing) via the expression of multiple guide RNAs [13,14].
Earlier approaches to gene editing
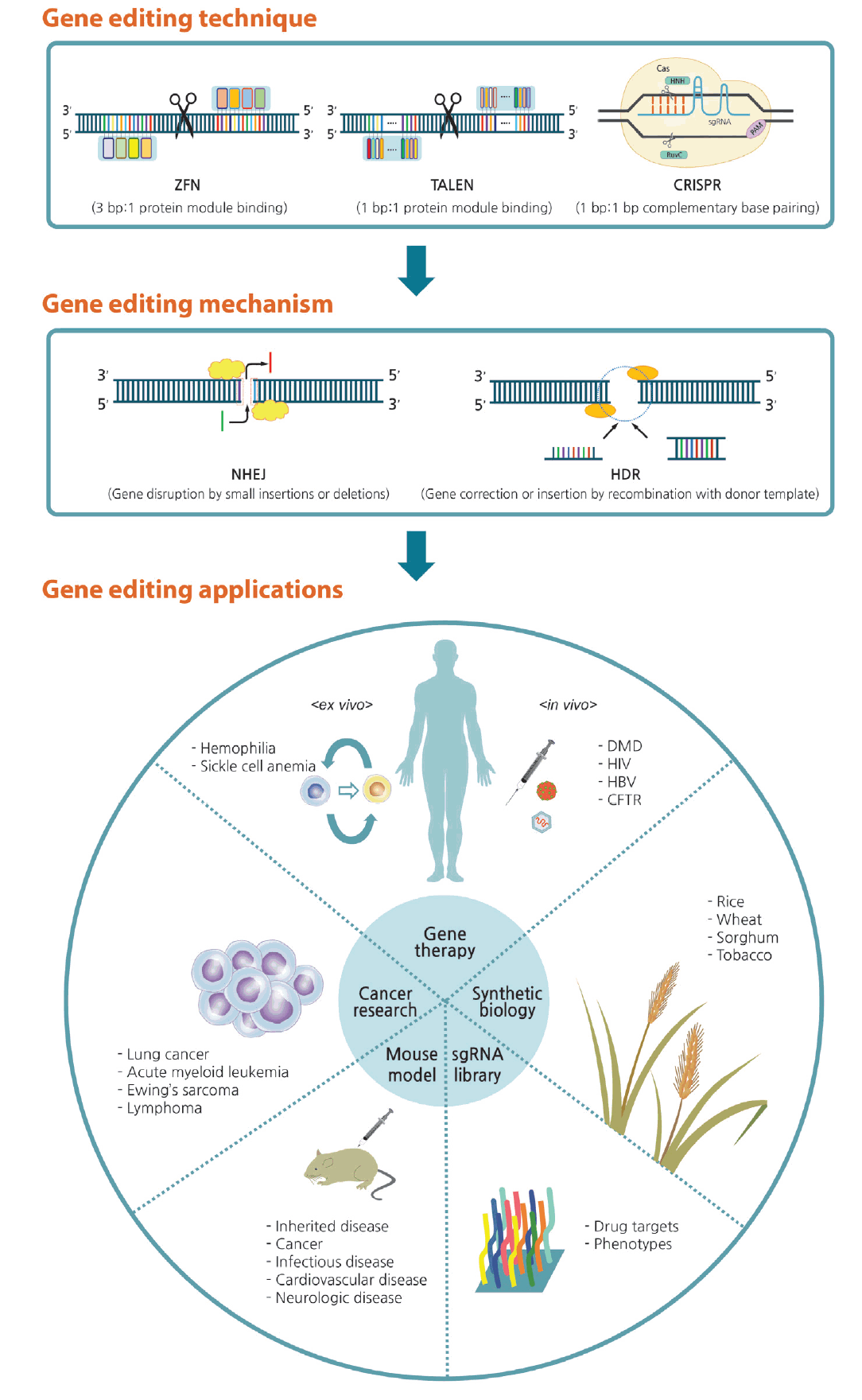
Prior to the advent of CRISPR technology, biologists used several generations of tools, all of which employed site-specific DNA DSBs for genome editing. To date, four types of DNA-binding nuclease have been developed: meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the most recently identified nuclease, Cas9.
Each of the previous tools had unique limitations. Meganucleases are like restriction enzymes but are programmed to target DNA sequences 14 to 40 bp in length. Owing to several shortcomings, including their lack of specificity in DNA recognition and the need to fuse the recognition and cleavage domains, meganucleases were used only briefly [47]. ZFNs and TALENs function according to similar principles, but differ in that their binding domains consist of three- and one-nucleotide recognition modules, respectively. These enzymes have separate DNA-binding domains and nonspecific cleavage domains, namely, FokI endonucleases, making them more efficient than meganucleases [48,49].
However, the construction of ZFNs remains a challenge due to the need to account for context-dependent binding preferences [50], notwithstanding previous efforts to circumvent this shortcoming. TALENs, despite having the advantage of one-to-one binding between the Transcription activator-like effector (TALE) unit and each base pair [51], require painstaking molecular biology cloning methods to synthesize highly conserved and repetitive TALE structures [52]. Consequently, the comparatively facile protein engineering of CRISPR makes this approach much more affordable and practical compared with precursor technologies (Table 1).
PART 2. APPLICATIONS OF CRISPR
Basic application of the CRISPR-Cas9 system
When DSBs are introduced, the lesion may be corrected by one of two major repair pathways: homology-directed repair (HDR) or nonhomologous end joining (NHEJ). HDR allows the exchange of genetic information between DNA molecules with similar sequences, whereas NHEJ forms short insertions or deletions (indels) in the target sequence. NHEJ does not require a repair template, but the resultant indels can cause frameshift mutations that lead to the production of nonfunctional, incomplete proteins, or to micro-RNA degradation by nonsense-mediated decay. On the other hand, the HDR machinery can repair DNA using exogenous single- or double-stranded DNA templates with sequence similarity to the DSB site. Thus, exploitation of HDR has allowed researchers to insert new genetic information at a target site, or to perform direct replacement of a mutated gene [45,46].
Although this approach was revolutionary, the natural process of HDR is inefficient because it requires selection and screening to identify the one-in-a-million cell in which homologous recombination has exchanged the wild-type gene for the desired modified version. However, CRISPR-Cas9 technology allows the inducible formation of DSBs, so that scientists can modify gene expression at the repair sitex; thus, opening a new avenue in genome editing [42].
Cell-based and in vivo animal studies
The applications of CRISPR-Cas9 have expanded into fields such as agricultural products, livestock, disease modeling, and therapeutics. In this section, we focus on the therapeutic aspects of gene-based diseases, especially monogenic disorders (Fig. 4).
In gene therapy, genes in diseased cells and tissues can be corrected by two approaches: ex vivo and in vivo editing [46]. In ex vivo therapy, the target cell population is removed from the body, modified using a programmable nuclease, and then transplanted back into the original host; thereby, preventing complications due to immunological rejection. By contrast, in vivo editing therapy involves direct transfer of genome-editing reagents, such as a programmable nuclease and donor templates, into the human body [53]. Each approach has advantages and disadvantages, and they are implemented differently to treat particular disorders. There has been examples of gene-editing techniques applied in disease cell lines (Table 2) [54-77] and in disease mouse models (Table 3) [60,63-66,78-88]. Furthermore, scientists have reported series of therapeutic applications with genome editing using stem cell (Table 4) [89-111].
Inactivation or correction of deleterious mutations
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is the most prevalent fatal genetic disease passed on through the X chromosome. The gene dystrophin consists of 79 exons, and several types of mutation in exon sequences lead to DMD. Currently, there is no effective treatment for DMD, but genome editing has the potential to restore expression of a modified dystrophin gene [53].
Efforts aimed at correction of the dystrophin gene in immortalized patient myoblasts with ZFNs and TALENs were initiated in 2013 [54,55]. Because 13% of DMD patients have a mutation in exon 51, the introduction of indels into, or complete excision of, exon 51 can restore dystrophin expression [56]. In one study, permanent removal of exons 45 to 55 by multiplexed Cas9 was therapeutically applicable in 62% of patients [57].
Mouse models can provide data that is relevant to in vivo human therapy. For example, the Mdx model mouse harbors a mutation in exon 23 of the dystrophin gene. Local and systemic delivery of gene correction to Mdx mice using adeno-associated virus (AAV) vector and the CRISPR-Cas9 system resulted in 2% to 100% correction, and the therapeutic benefits were predicted to be 15% to 20% [78-81].
Genome editing has also been effective in DMD gene therapy in patients lacking exon 44; in this case, the correction was performed ex vivo in induced pluripotent stem cells (iPSCs). Three correction strategies were tested: skipping of exon 45, introduction of small indels resulting in a frameshift in exon 45, and knock-in of the missing exon 44 to restore the full protein coding region; the last of these strategies was the most effective. The corrected iPSCs successfully differentiated into muscles and expressed functional protein [89].
Insertion of corrective or protective mutation
Hemophilia
Hemophilia is caused by different genetic mutationsŌĆö in coagulation factor VIII (F8) for hemophilia A, and in coagulation factor XI (F9) for hemophilia B. Gene therapy is an option for treating hemophilia because correction of the defective gene results in permanent expression of functional protein, and even 1% wild-type expression of coagulation factor VIII or XI is sufficient to confer a therapeutic effect [112,113].
The first successful in vivo gene targeting of hemophilia was achieved in a hemophilia B neonate mouse [82]. Using a ZFN pair to target the defective human F9 (hF9) gene, and AAV as the delivery vector, donor cDNA was inserted into the mouse genome. A similar approach in adult hF9 mice resulted in stable production of human factor IX [83].
Hemophilia A, which is more prevalent than hemophilia B, involves a more complex type of mutation, making it harder to edit the gene. However, chromosomal inversion at the F8 gene is a common cause of hemophilia, and TALENs were previously shown to be able to correct this rearrangement [90]. Thus, the CRISPR-Cas9 system was used to target each side of the ~600 kb inversion and correct the mutation in iPSCs derived from hemophilia A patients [91].
Sickle-cell anemia and ╬▓-thalassemia
Sickle-cell anemia and ╬▓-thalassemia are both caused by mutations in the HBB gene, resulting in an inappropriate level of the ╬▓-globin chain of hemoglobin. Editing the ╬▓-globin locus by targeted nucleases represents a new strategy for permanently curing hemoglobinopathies. TALENs programmed to target the ╬▓-globin locus were used for HDR-mediated full-length cDNA knockin in K562 cells [58], and ZFNs were used to correct a sickle-cell anemia-associated point mutation in CD34+ hematopoietic stem progenitor cells [92]. These hemoglobinopathies are particularly advantageous for gene therapy because extracted patient iPSCs can be differentiated into hematopoietic stem cells, which can in turn be inserted back into patients by autologous transplantation. This strategy has already been implemented with all ZFNs, TALENs, and CRISPR-Cas9 in both sickle-cell anemia and ╬▓-thalassemia patients [93-97].
Disruption of viral DNA
Human immunodeficiency virus
The most advanced gene-editing strategy is the ex vivo modification of T cells to knock out the cinnamoyl-CoA reductase 5 (CCR5) gene, resulting in resistance to human immunodeficiency virus (HIV) infection. This is one of the few cases in which a treatment that exploits the gene-editing machinery has been used in clinical trials (Table 5). This idea was clinically validated when an HIV-infected patient received a stem cell transplant from a donor with a homozygous deletion in the CCR5 allele, resulting in undetectable levels of HIV and restoration of normal CD4+ T cell counts [114].
One study demonstrated the safety of infusion of ZFN-modified autologous CD4+ T cells bearing a deletion of the CCR5 gene into HIV-positive human patients [115]. Building on the results obtained with ZFNs, multiple efforts were made using similar gene-editing strategies to knock out CCR5 using TALENs or CRISPR-Cas9. Wild-type iPSCs were seamlessly modified by NHEJ-mediated deletion of the CCR5 gene, at an average rate of 14% with TALENs and 33% with CRISPR [98]. To increase resistance to HIV infection, other genes were targeted in addition to CCR5, including C-X-C chemokine receptor type 4 (CXCR4), which encodes a coreceptor, and PC4 and SFRS1 interacting protein 1 (PSIP1), which encodes the lens epithelium-derived growth factor (LEDGF)/p75 protein required for HIV integration [99,116,117].
To reduce adverse off-target effects, several studies have attempted to eliminate the integrated HIV-1 genome. In one study, for example, the HIV-1 long terminal repeat (LTR) U3 region was efficiently targeted by the CRISPR-Cas9 system, resulting in inactivation of viral gene expression and replication in CHME5, HeLa, TZM-b1, and U1 cells [59].
Beyond HIV, programmable gene-editing nucleases have also been applied to other viral pathogens. For example, in Huh7, HepG2, HepAD38, and HepaRG cells transfected with a hepatitis B virus (HBV) expression vector, targeted editing of multiple genes was used to reduce the production of HBV core and surface proteins. HBV-expressing templates were disrupted by CRISPR-Cas9 both in vitro and in vivo [60-66]. Likewise, the E6 and E7 genes of human papillomavirus were also targeted by CRISPR-Cas9 [67-70].
There are increasing numbers of ongoing and completed clinical trials adopting gene-editing technology (Table 5).
PART 3. FUTURE PERSPECTIVES
Cancer research
All cancers harbor multiple mutations that cause cells to grow progressively and express malignant phenotypes. These mutations can be categorized into four types: oncogenes, tumor suppressors, epigenetic factors and control loci, and chemoresistance genes. The CRISPR-Cas9 system represents a powerful, highly specific and adaptable tool for correcting such mutations and treating the cancers that contain them [118]. While oncogenic changes occur in many cancers and play important roles in malignant cell proliferation, oncogenes such as the receptor tyrosine kinase Erb2 can be targeted directly by CRISPR-Cas9 [119].
From another perspective, it is possible to utilize CRISPR-Cas9 to introduce cancer-causing mutations in human cell lines and animal models. In this context, the following cell lines have been constructed to date: lung cancer [120], acute myeloid leukemia [121], liver cancer [122], and pancreatic cancer [123].
Animal models
CRISPR-Cas9 technology can be applied to animal models for the study of both cancers and other inherited diseases. Heritable gene modification can be achieved by injecting CRISPR-Cas9, targeting one or multiple alleles, directly into fertilized zygotes [42]. Among transgenic animal models, mice are the most widely used in experiments because of the relatively short time required to generate mutants; however, non-human primate models have been created successfully by multiplex gene targeting, potentially generating superior systems for the study of complex human diseases, for example, neurodegenerative disorders [124]. Nonetheless, mouse models remain the most cost-effective. Moreover, mice are amenable to large-scale in vivo mutagenesis studies, especially when highly specific targeted editing can avoid the confounding effects of off-target mutagenesis [125].
Synthetic biology
The applications of the CRISPR-Cas9 system to synthetic biology include all concepts related to synthetic gene circuits in living cells. Because synthetic gene circuits consist of sensors, processors, and actuators, synthetic biology has the potential not only to advance basic research, but also to enable practical applications in medicine, biofuel production, and synthesis of commodity chemicals [126]. The most practical applications of the CRISPR-Cas9 system have been in plants, especially crops such as rice, wheat, sorghum, and tobacco. For example, CRISPR-Cas9 was used to target and knock out the mildew-resistance locus (MLO) genes, which encode proteins that repress the defense against powdery mildew disease in hexaploid bread wheat [127].
sgRNA library
CRISPR-Cas9 can also be applied to the systematic analysis of gene functions in human cells. A lentiviral sgRNA library was developed against genes identified by functional screening and high-throughput sequencing analysis. This powerful loss-of-function library screening is expected to facilitate discovery of genes that participate in various biological processes, including drug targeting, toxicity, and expression of certain phenotypes [128].
Induced pluripotent stem cells
iPSCs, which are very similar to embryonic stem cells, are pluripotent cells with a high self-renewal rate that can differentiate into almost all cell types; however, their utilization is associated with significantly less ethical controversy than that of their embryonic counterparts. Recent advances in stem cell technology are likely to provide great benefits to the clinical use of iPSCs in clinical applications [129].
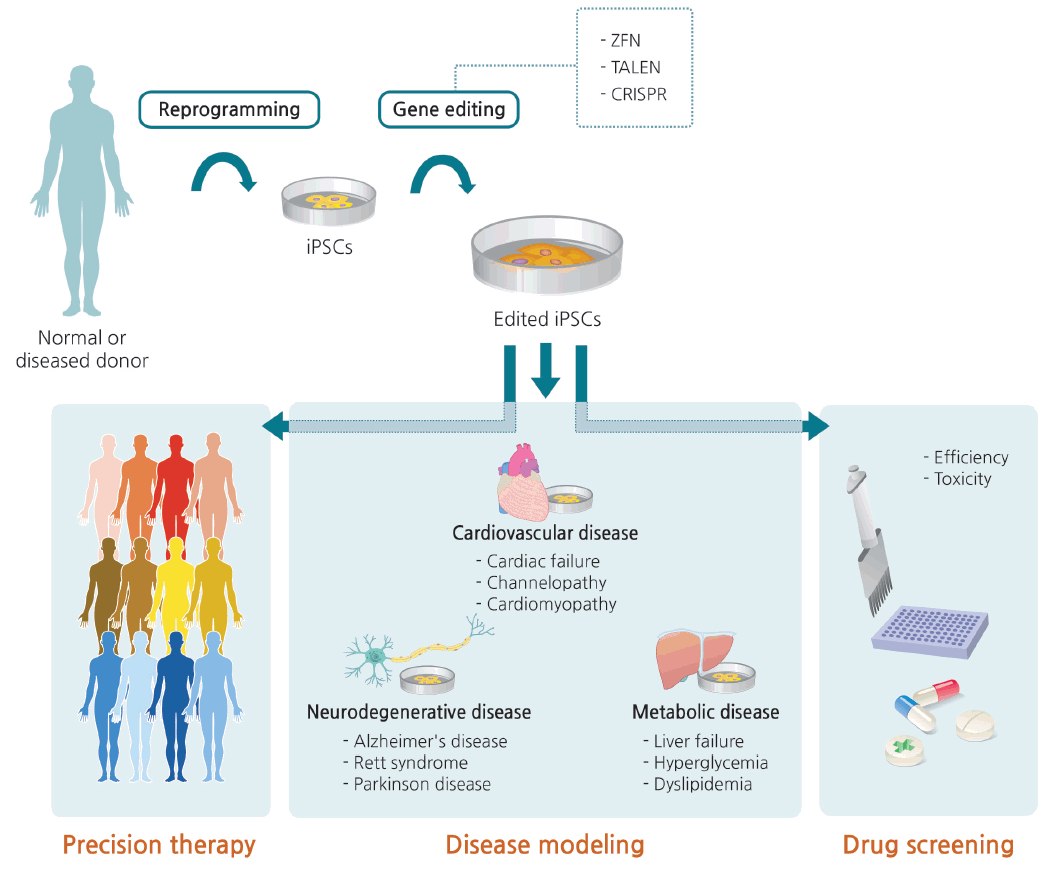
As mentioned above, iPSCs have a major advantage for personalized medicine because they can be derived from the patients themselves, and can therefore avoid immune rejection when transplanted. Ex vivo therapy includes correction of patient-derived iPSCs through gene editing, as well as differentiation into nonrenewable cell types such as neurons and cardiomyocytes (Fig. 5) [100].
In addition to this type of precision therapy, human iPSC lines with genotypes characteristic of specific diseases could be used to understand pathogenic mechanisms. Disease modeling and drug efficiency/toxicity testing with iPSCs not only increase the accuracy of disease simulation, but are also less expensive than generating animal models. However, care must be taken when interpreting the results of phenotypic comparisons between patient iPSC-derived cells and healthy control cells. Specifically, the results are vulnerable to confounding variables that might influence the phenotypes of interest, including epigenetic status and unmatched age, gender, and ethnicity. In this respect, gene editing is the only way to distinguish changes that are specifically relevant to a given disease [130].
The CRISPR-Cas9 system enables simultaneous knockout of multiple genes, as well as knock-in of specific alleles in iPSCs, distinguishing it from earlier gene-editing technologies such as ZFNs and TALENs. An isogenic human iPSC cell line precisely corrected by the CRISPR-Cas9 system was recently constructed, despite the handling difficulties associated with gene editing of human stem cells [131]. In the future, the use of CRISPR-Cas9 with iPSCs will lead to novel combinations of gene and cell therapies [132].
Areas of technical improvement: DSB repair, nucleases, delivery
Prior to the clinical application of CRISPR-Cas9 in human patients, the safety and efficacy of the system must be validated. The specificity and efficiency of genome-editing tools can be improved by targeting DSB repair pathways, modifying nucleases, and changing the mode of delivery. We briefly discuss each topic below.
DSB repair
As noted previously, DNA editing rates are currently determined by the two major endogenous DSB repair pathways. HDR is more suitable for gene correction or gene insertion than NHEJ, which creates indels that generally induce a loss of function. NHEJ is more efficient because it is active throughout the cell cycle and does not require a repair template; by contrast, the rate of HDR is inherently low. In addition, some HDR components are expressed only during the S/G2 phase, limiting the use of HDR-based editing approaches to dividing cells and preventing their use in post-mitotic cells, such as neurons and cardiac myocytes. For this reason, controlling the efficiency of HDR has become a major focus of efforts aimed at increasing the effectiveness of gene correction [46].
One strategy for improving the efficiency of HDR is the suppression of NHEJ during DSB repair. Suppression of a key enzyme in the NHEJ pathway increases the efficiency of HDR-mediated genome editing up to 19-fold [133,134]. Another strategy involves the induction of HDR-like corrections in post-mitotic cells via a novel non-HDR-based pathway: microhomology-mediated end joining [135,136]. Using microhomologous sequences (5 to 25 bp), the so-called PITCH (Precise Integration into Target Chromosome) system can produce precise gene knock-ins. Meanwhile, the lower success rate of CRISPR-Cas9 relative to TALEN could be overcome by the generation of sticky, instead of blunt, ends [137].
Nucleases
From a clinical standpoint, highly specific gene engineering technology is essential because specificity is correlated with safety. Unexpected off-target mutations may cause cells to become carcinogenic or functionally impotent. Because genetic modifications are permanent, multiple ongoing research efforts are devoted to the reduction of off-target effects.
One strategy for achieving this goal is to improve the targeting specificity of Cas9. Careful design of sgRNA, including avoidance of poly-G/poly-C-rich targets, as well as tight control of the amount and duration of Cas9 and sgRNA expression, are both important for high specificity [138]. The use of modified Cas9 with two separate sgRNAs that each generate a single-strand nick on opposing DNA strands can potentially reduce off-target activity by 50- to 1,500-fold in cell lines [139,140]. Additionally, truncation of the guide RNA, with a target-complementary region shorter than 20 nucleotides in length, can decrease off-target activity by 5,000-fold or more [141]. Moreover, a fusion protein of catalytically inactive Cas9 and FokI nuclease (fCas9) can recognize the target DNA site with 140-fold greater specificity than the wild-type protein in human cells [142,143].
Delivery
The delivery of gene-editing tools to target cells is another major challenge with respect to efficacy and specificity. Both viral and nonviral delivery methods are currently being evaluated for the introduction of Cas9 into target cells ex vivo or in vivo. Depending on the mode of delivery and the duration of nuclease expression, both off-target activities and immune reactions are possible.
Viral vectors, such as AAV or lentivirus, represent the most common delivery systems, and these vectors have recently been approved for clinical use. In particular, AAV, which was recently clinically approved, is an attractive candidate for in vivo use because of its low degree of immune stimulation, well-characterized serotypes, and ability to target diverse tissues such as eye, brain liver, and muscle [144]. However, one challenge of using AAV is its relatively small packaging capacity (4.7 kb). Consequently, efforts are underway to deliver the Cas9 and sgRNA coding sequences using two separate AAV vectors. Alternatively, these size constraints can be sidestepped by creating a shorter Cas9 ortholog. In addition to size incompatibility, viral vectors have the drawback of possible constitutive nuclease expression, resulting in cell toxicity and genomic instability [145].
A variety of nonviral methods exist for both in vivo and ex vivo delivery of CRISPR-Cas9 in the form of mRNA or proteins [146]; these include electroporation, hydrodynamic delivery, and the use of liposomes [147]. Because mRNA and proteins introduced by these methods are present in cells only transiently, nonviral delivery systems are expected to decrease the frequency of off-target effects and cell toxicity relative to viral systems.
Ethics
As promising as CRISPR sounds, a variety of concerns have been expressed about this technique. In early 2015, a Chinese research group used CRISPR-Cas9 to perform editing on nonviable human trinuclear zygotes, stimulating a vigorous discussion of the ethical implications. The International Summit on Human Gene Editing in Washington was convened in late 2015, providing a forum in which scientists could achieve a consensus on human germline engineering.
The CRISPR-Cas9 toolbox has many advantages and it can be used to correct many defects that occur systemically or from birth, including cystic fibrosis and HuntingtonŌĆÖs disease. However, it remains unclear how we should set boundaries regarding which human traits are appropriate for editing. In addition, few would argue that a number of safety and efficacy concerns about CRISPR-Cas9 remain to be resolved. Especially in light of the possibility that undesirable parties could use this technology for eugenics, it would be irresponsible to allow modification of human embryos. In this respect, we must confront the need for robust regulation, even as arguments rage between the advocates of caution and progress.
CONCLUSIONS
Recent advances in genome editing with CRISPR is very rapid from basic research to clinical therapy. Development of CRISPR may widen the opportunity of iPSC application in real clinic. To deeply understand cutting-edge CRISPR technology can promote gene-editing applications in fields of cancer research, synthetic biology, and gene therapy using induced pluripotent stem cells.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





