


INTRODUCTION
Gastric cancer (GC) remains one of the most prevalent and lethal malignancies worldwide and the fifth leading cause of cancer-related deaths [1]. Despite advancements in treatment, the prognosis of patients with GC remains poor, particularly for those diagnosed at advanced stages [2]. Early detection of GC, ideally during its premalignant stages, is critical for improving the prognosis and reducing the global burden of the disease. Although endoscopic inspection remains the mainstay of screening and diagnosis for GC, this method is invasive, causes patient discomfort, and increases healthcare costs [3]. However, current non-invasive screening and diagnostic methods for GC lack the sensitivity and specificity necessary for early stage detection, underscoring the need for novel biomarkers and innovative approaches [4].
Emerging evidence suggests that the microbiome plays a crucial role in the pathogenesis of GC [5]. Helicobacter pylori is a well-established and unmatched risk factor implicated in the development and progression of GC. Recent research suggests that alterations in the gastric microbiome other than H. pylori may play a role in disease progression from chronic gastritis to atrophic gastritis, intestinal metaplasia, dysplasia, and eventually carcinoma [6]. These findings highlight the potential of microbial signatures as biomarkers for early detection of GC.
Microbial-derived extracellular vesicles (EVs), which are nanosized particles secreted by various microorganisms, represent a promising frontier for exploring the role of the microbiome in cancer. These vesicles carry proteins, lipids, nucleic acids, and other molecules that reflect the biological state of their parent cells, and play a significant role in cell signaling by acting as carriers of molecular information between cells [7]. Importantly, microbial-derived EVs are present in various biological fluids, such as saliva, serum, urine, and gastric juice, making them accessible targets for noninvasive diagnostic testing. EVs have attracted significant interest as potential biomarkers for various diseases, including cancer, owing to their ability to protect and transport microbial components in a stable form [8].
In this study, we analyzed microbially derived EVs from various liquid biopsy samples obtained from patients at different stages of gastric carcinogenesis. We aimed to explore the microbial features associated with GC using microbial-derived EVs and to assess whether these features vary across disease stages. Additionally, this study aimed to deepen our understanding of the role of microbes in GC progression and potentially inform the development of future biomarkers for disease detection and staging.
METHODS
Study participants and sample collection
Participants were prospectively recruited at the Chung-Ang University Hospital between December 2016 and October 2022. The inclusion criteria for the neoplasm group were newly diagnosed patients with histologically confirmed GC or adenoma. The control group included patients with only gastritis and no evidence of gastric neoplasms on endoscopic examination. The exclusion criteria included individuals with a history of malignancy (including GC), those who had undergone gastric surgery, those who had taken antibiotics or probiotics within the past three months, or those under 20 years of age. The final cohort comprised 141 participants, including 132 patients and nine controls (100 male and 41 female). This study was approved by the Institutional Review Board (IRB) of the Chung-Ang University Hospital (IRB No. C2016047(1790)) and adhered to the Declaration of Helsinki. The informed consent was provided in written form by all participants. Gastric neoplasms were diagnosed and categorized using the Vienna Classification System [9]. Specifically, category 3 (non-invasive low-grade adenoma/dysplasia) was classified as low-grade dysplasia (LG), category 4.1 (high-grade adenoma/dysplasia) and 4.2 (non-invasive carcinoma [carcinoma in situ]) were classified as high-grade dysplasia (HG), and categories 4.3 (suspicion of invasive carcinoma) and 5 (invasive neoplasia) were classified as GC.
The following protocols were used for sample collection. For saliva samples, participants were instructed to swish 10 mL of saliva in their mouths and then expectorate into a specimen tube, which was stored at -80°C until use. Gastric juice samples were collected following a specific protocol. Patients fasted for > 8 hours prior to collection. For those undergoing endoscopic examination or resection, a trap tube was attached between the endoscope and the suction tube. Gastric juice (7–30 mL) was suctioned through the endoscope at the beginning of the procedure. Simultaneously, tissue samples from the antrum and body were collected and stored at -80°C. For patients undergoing surgical gastrectomy, gastric juice and tissue samples were obtained either endoscopically before surgery or during surgery, immediately after making an incision. The endoscopes were washed and disinfected by immersion in a detergent solution containing 7% proteolytic enzyme and 2% glutaraldehyde to prevent contamination. Gastric juice samples were immediately stored at -20°C and transported to the laboratory without preservative reagents. The gastric juice was then diluted to 40 mM with 1 M Tris base and centrifuged at 600 ×g for 10 minutes at 4°C to obtain a supernatant, which was further centrifuged at 1,500 ×g for 10 minutes at 4°C. Finally, the supernatant was collected and stored at -80°C.
DNA extraction and sequencing
Bacterial EVs were isolated from the gastric juice, saliva, serum, and urine of the individuals following a previously described procedure [10,11]. Each gastric juice, saliva, and urine sample was centrifuged at 10,000 ×g for 10 minutes at 4°C. The supernatant was taken and passed through a 0.22 μm membrane filter to eliminate foreign particles and then quantified based on protein concentration [12,13]. For serum, after mixing 100 μL of serum and 900 μL PBS, it was centrifuged at 10,000 ×g for 10 minutes at 4°C to eliminate other components. The supernatant was taken and passed through a 0.22 μm membrane filter to eliminate foreign particles, and it was quantified based on protein concentration.
Bacterial DNA was extracted from the prepared EVs as previously described [11,14]. Isolated EVs (1 μg by protein, each sample) were boiled at 100°C for 40 minutes and centrifuged at 13,000 ×g for 30 minutes, and the supernatants were collected. The collected samples were then subjected to bacterial DNA extraction using a DNA extraction kit (PowerSoil DNA Isolation Kit; MO BIO, Carlsbad, CA, USA) following the manufacturer’s instructions. The isolated DNA was quantified using the QIAxpert system (QIAGEN, Hilden, Germany). Microbial genomic DNA was extracted using a DNeasy PowerSoil kit (QIAGEN) according to the standard protocol provided in the manufacturer’s instructions. The isolated genomic DNA was amplified by targeting the V3–V4 hypervariable regions of the 16S rRNA gene. This amplification was carried out using specific primers (16S_V3_F: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′ and 16S_V4_R: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′). Amplicon libraries were prepared from the amplified DNA fragments. The MiSeq Reagent Kit v3 (600-cycle) (Illumina, San Diego, CA, USA) was used to sequence the library reagents. The Nextra XT Index Kit v2 Set A (96 indices, 384 samples) (Illumina) was used for barcodes and adapters. Library preparation for sequencing followed the 16S Metagenomic Sequencing Library Preparation (Part # 15,044,223 Rev. B). All amplicons were sequenced using MiSeq (Illumina) according to the manufacturer’s instructions. The bacterial DNA in each sample was quantified using a QIAxpert system.
Taxonomic classification and microbial profiling
Paired-end 16S rRNA gene sequences were processed using the Quantitative Insights into Microbial Ecology (QIIME2 v2021. 4) [15]. Adapter sequences were trimmed using Cutadapt, and the reads were subsequently filtered for quality and the presence of chimeras using DADA2, with specific parameters set as follows: trim-left-f 0, trim-left-r 0, trunclen-f 260, trunc-len-r 200, trunc-q 2, max-ee-f 3, and maxee-r 3 [16]. A naïve Bayes classifier, trained on the V3–V4 region sequences from the SILVA 138 database, was used for taxonomic classification. Sequences identified as chloroplasts or mitochondria were excluded from the analysis.
Statistical analysis
For alpha diversity analysis, the samples were rarefied to a minimum read count of 2,386 to normalize the data. The p values for alpha diversity were calculated using analysis of variance adjusted for age and sex and were further adjusted using the bootstrap method [17]. Principal coordinate analysis (PCoA) was performed to evaluate the taxon-level clustering of groups based on Bray–Curtis dissimilarity distances. The synthetic minority oversampling technique (SMOTE) algorithm was applied to increase the normal sample size for PCoA [17]. The p value for the PCoA was determined through permutational multivariate analysis of variance using dissimilarity matrices adjusted for age and sex. For the analysis of relative abundances and Multivariable Associations with Linear Models2 (MaAsLin2), operational taxonomic units (OTUs) that exhibited a low count across the dataset were filtered out; specifically, OTUs with a minimum count of less than or equal to four were removed [18]. Furthermore, samples were retained only if they contained OTUs that surpassed the minimum count threshold in more than 20% of the dataset, thereby ensuring the exclusion of samples with sparse OTU representation. To address the low variance, which often indicates a lack of differential abundance across conditions, we implemented a filtering process based on the interquartile range. Features within the lowest 10% of the interquartile range were discarded. Following the filtering process, data normalization was performed to mitigate the effects of varying sequencing depths across the samples. Total sum scaling was used as the normalization technique, where the read count of each sample was divided by the total read count of the sample to ensure comparability across the dataset. The MaAsLin2 method was used to evaluate differences in the OTU counts of bacteria-derived EVs among groups. This analysis was performed using a negative binomial model for group comparisons. Analysis was conducted using MaAsLin2 on MicrobiomeAnalyst (https://www.microbiomeanalyst.ca/) and the MaAsLin2 package in R adjusting for age and sex as covariates. Statistical significance was set at a q-value of less than 0.05. MaAsLin2 was executed using the default parameters in conjunction with the Benjamini–Hochberg false discovery rate method [19]. To validate and complement the results of MaAsLin2, a random forest classifier analysis was performed. To balance the sample sizes across groups, SMOTE oversampling was applied, increasing the sample size of each group (control, LG, HG, and GC) to 100 samples. A random forest classifier was subsequently employed to distinguish between the disease groups. The dataset was split into a training set (70%) and a test set (30%) to rigorously assess the model’s predictive accuracy. For feature selection, microbial biomarkers identified using MaAsLin2 were incorporated into the random forest model. These biomarkers were selected based on their statistically significant associations with the disease groups, serving as key input variables to improve the classifier’s interpretability and overall performance. All statistical analyses were performed in R Software (version 3.6.1; R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Clinical characteristics of the study population
A total of 141 participants were included in the study, including nine controls, 58 with LG, 33 with HG, and 41 with GC. We evaluated samples from four sources: gastric juice, saliva, serum, and urine. A total of 550 samples of all types and conditions were analyzed. The mean age of the study participants was 64.4 years, with males accounting for 70.9% (100 out of 141) of the study population. Table 1 summarizes the clinical characteristics of the enrolled patients, categorized by sample type.
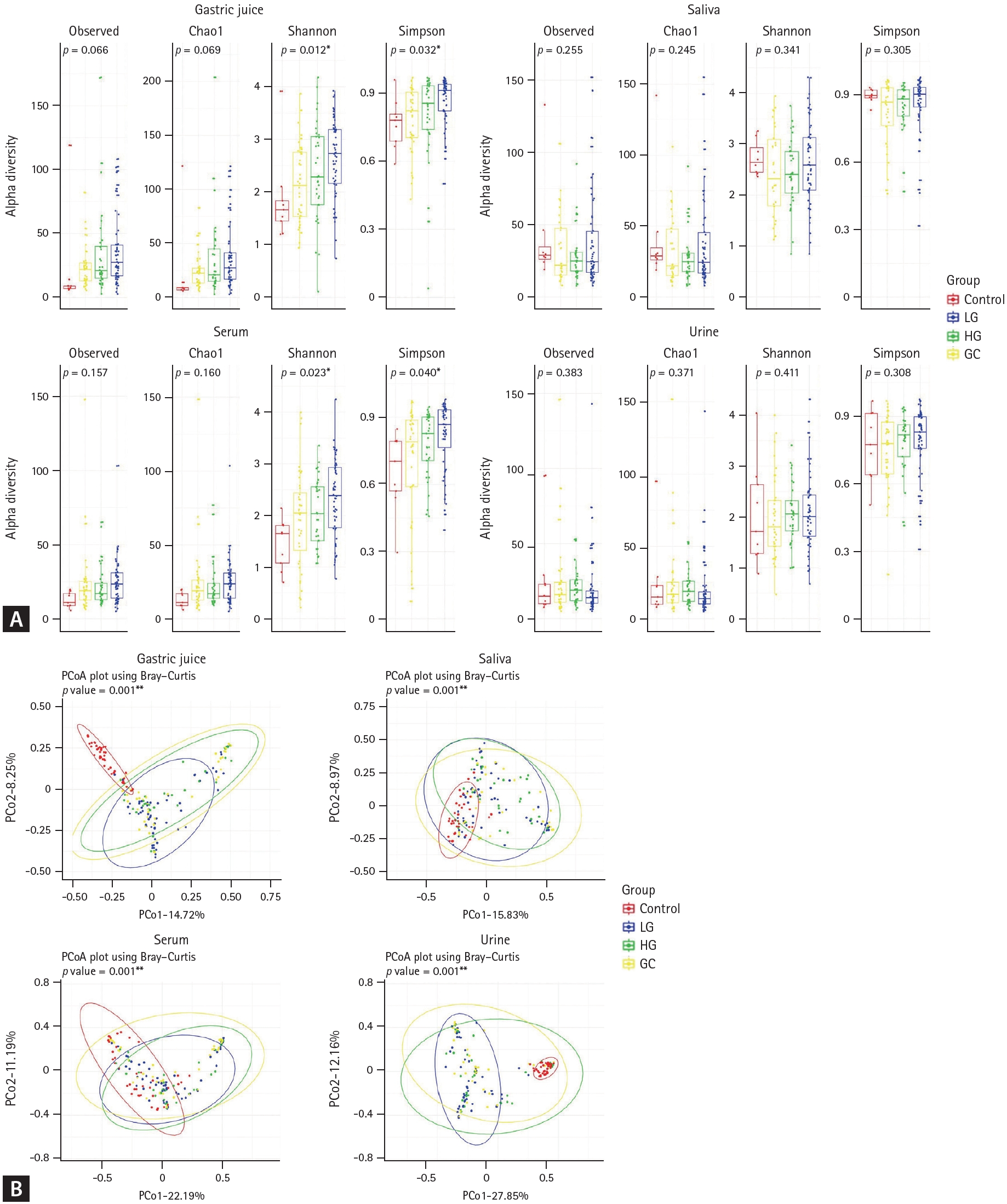
Diversity of microbial communities in EV samples
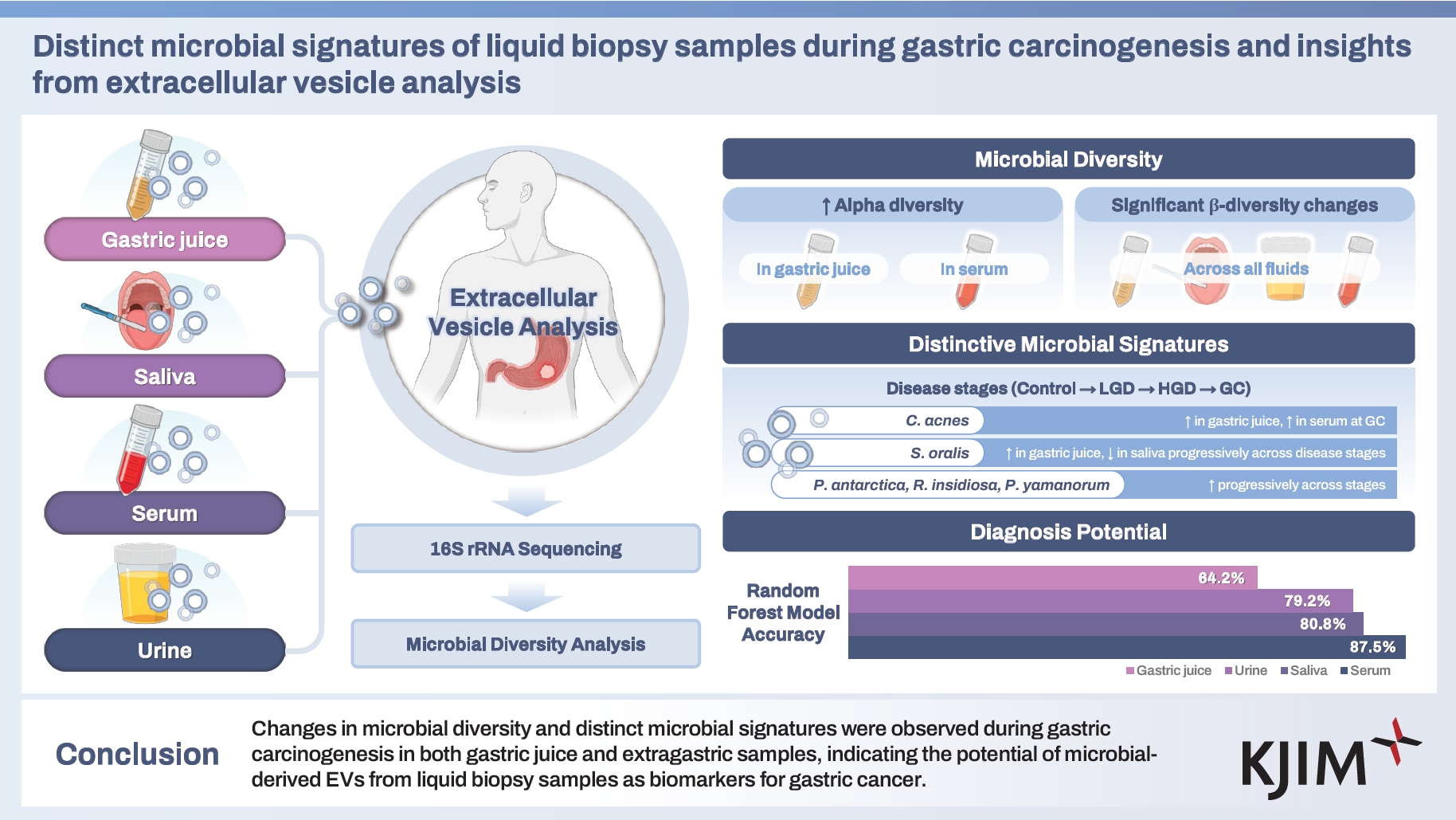
The alpha diversity was assessed in the gastric juice, saliva, serum, and urine samples using various indices (Fig. 1A). In gastric juice, both the Shannon (p = 0.012) and Simpson (p = 0.032) indices showed significant changes in microbial diversity and species dominance with the progression of the pathological condition. Similarly, in the serum, significant changes in microbial diversity were observed in Shannon (p = 0.023) and Simpson (p = 0.040) indices as the disease progressed. In contrast, the saliva and urine samples did not show statistically significant differences in microbial diversity across the different stages of gastric carcinogenesis.
Beta diversity was assessed across different stages of gastric carcinogenesis in all sample types. The results showed significant differences in microbial composition across all sample types during disease progression (p < 0.05, Fig. 1B), with notable differences in gastric juice and urine (p < 0.001, Fig. 1B).
Alterations in microbiota across different disease stages in various samples
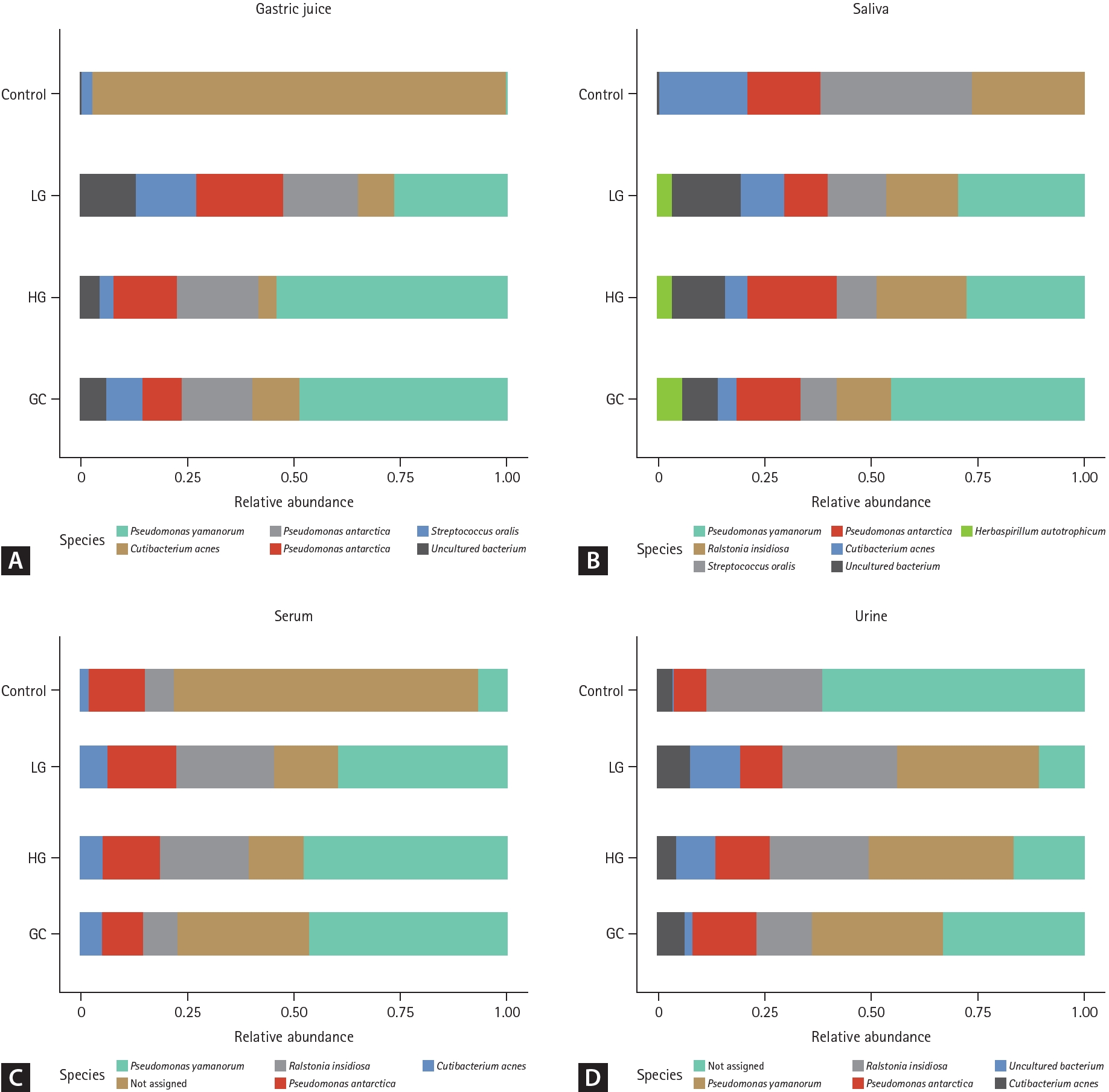
The relative abundances of bacterial communities in various samples (gastric juice, saliva, serum, and urine) were analyzed proportionally, as shown in Figure 2. The bar chart represents the relative abundances of bacterial species identified in the four groups: control, LG, HG, and GC.
In gastric juice samples (Fig. 2A), Cutibacterium acnes dominated in the control group, whereas in the LG, HG, and GC groups, there was a decrease in the abundance of C. acnes and an increase in the abundances of Pseudomonas yamanorum, Pseudomonas antarctica, Ralstonia insidiosa, and Streptococcus oralis. For saliva samples (Fig. 2B), compared with the controls, the LG, HG, and GC groups showed a decrease in C. acnes and S. oralis abundances, along with an increase in P. yamanorum abundance. Notably, Herbaspirillum autotrophicum was detected exclusively in the disease groups (LG, HG, and GC). In the serum samples (Fig. 2C), compared to the controls, the LG, HG, and GC groups showed an increase in C. acnes and R. insidiosa abundances. Urine samples (Fig. 2D) revealed a heterogeneous bacterial composition. However, P. yamanorum was consistently prominent across the disease groups (LG, HG, and GC), indicating a noticeable increase compared to that in the control group.
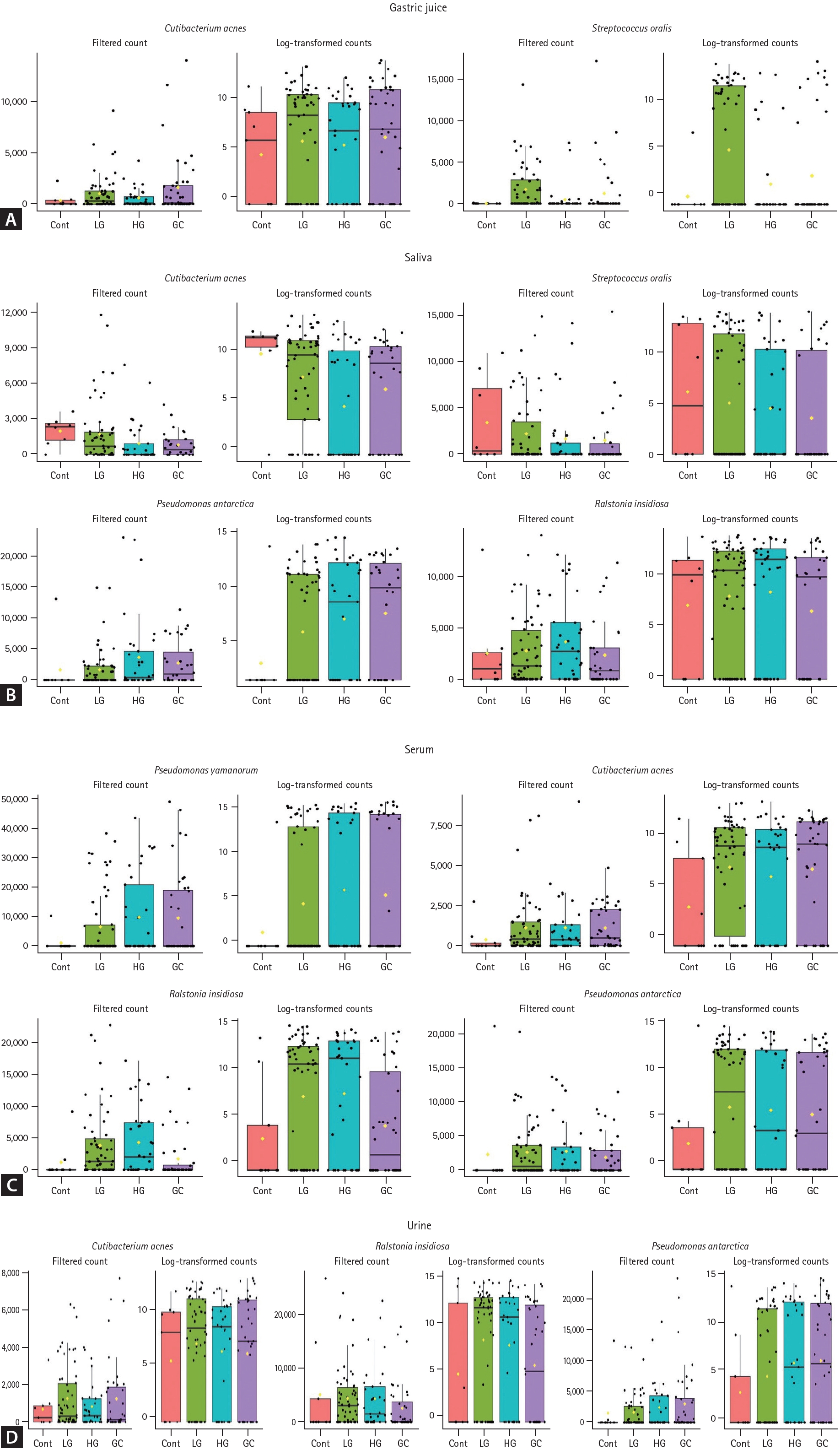
Differential bacterial abundance in biological samples across disease states
Specific bacterial strains with significantly different filtered OTU counts in the disease groups (LG, HG, and GC) compared to those in the control group were identified. Figure 3 shows a comparative analysis of the abundances of these bacterial strains across different patient groups for various sample types. Bacterial abundance was represented by filtered counts to assess the correlations between groups. In the gastric juice, the abundance of C. acnes and S. oralis displayed a notable increase with the progression of disease severity (q < 0.01 each, Fig. 3A). In saliva, the levels of C. acnes and S. oralis significantly decreased as the disease progressed, whereas P. antarctica and R. insidiosa increased (q < 0.01 all, Fig. 3B). In the serum, the abundances of P. yamanorum, C. acnes, R. insidiosa, and P. antarctica significantly increased with advancing disease stage (q < 0.01 all, Fig. 3C). Similarly, in urine, C. acnes, R. insidiosa, and P. antarctica showed significantly increasing trends during disease progression (q < 0.01 each, Fig. 3D). To complement the MaAsLin2 results and further assess the discriminative power of these identified bacterial biomarkers across disease stages, the random forest classifier was employed. This analysis evaluated the distribution and classification potential of specific microorganisms associated with different stages of gastric carcinogenesis in each sample type (Supplementary Tables 1-4). The overall classification accuracy was 64.2% for gastric juice and 80.8% for saliva, while serum and urine samples demonstrated classification accuracies of 87.5% and 79.2%, respectively. Meanwhile, it is well known that differences in microbiome diversity and composition are greatly influenced by H. pylori infection status. To assess the impact of H. pylori, all analyses were re-evaluated after excluding samples from H. pylori-positive participants. The overall trends and results remained consistent (data not shown).
DISCUSSION
This study aimed to investigate shifts in microbial communities across different stages of gastric carcinogenesis by analyzing microbial-derived EVs in various biological samples, including gastric juice, saliva, serum, and urine. We observed significant changes in microbial diversity and composition during gastric carcinogenesis, which were effectively captured through metagenomic analysis of samples obtained from liquid biopsy.
Our results demonstrated significant changes in the microbial diversity in gastric juice and serum samples as the disease progressed. The increased alpha diversity observed in gastric juice suggests greater microbial variation and shifts in species dominance associated with GC development. These findings are consistent with those of previous studies that highlighted the dynamic nature of the gastric microbiome in response to carcinogenic changes in the gastric environment [5]. Similar patterns of increased microbial diversity were observed in the serum samples, which may reflect systemic changes in the microbiome associated with advanced cancer stages. Conversely, saliva and urine samples did not show statistically significant changes in alpha diversity, suggesting that microbial alterations associated with GC may be more localized in the gastric environment and systemic circulation than in other bodily fluids. Although the alpha diversity in saliva remained relatively stable, significant changes were observed in gastric juice, which might reflect the selective pressure of the gastric environment during gastric carcinogenesis. These changes were particularly pronounced in the beta diversity analysis, which showed significant differences in the microbial community composition across all sample types during gastric carcinogenesis. These results support the hypothesis that GC progression is associated with specific shifts in microbial populations [20]. This finding aligns with emerging evidence suggesting that changes in the microbial community structure could serve as potential indicators of disease status and progression [21].
We further analyzed the microbial composition and abundance and identified distinct microbial signatures across different liquid biopsy samples during carcinogenesis. In gastric juice, the abundance of C. acnes and S. oralis significantly increased with disease progression, suggesting that these bacteria might be involved in the pathogenesis or progression of GC or that they may thrive in the altered gastric environment associated with the disease. In contrast, the abundances of C. acnes and S. oralis decreased in the saliva as disease severity increased. This opposing pattern may indicate different microbial dynamics in the oral and gastric environments during GC progression. C. acnes, P. antarctica, and R. insidiosa also increased in both the serum and urine, with the latter two species displaying a similar increasing trend in the saliva. Meanwhile, P. yamanorum exhibited a notable increase, specifically in the serum. These common microbial signatures across different samples may indicate the importance of these species in the process of carcinogenesis and their association with changes in the microbial milieu of the stomach. This consistency in microbial presence across various liquid biopsy samples suggests the possible systemic circulation of microbial-derived EVs, as proposed in previous studies [22]. These variations in microbial populations align with a growing body of evidence suggesting a complex interaction between the microbiota and cancer cells [23]. Furthermore, these findings provide a basis for developing diagnostic approaches using extragastric liquid biopsy samples such as serum and urine to detect microbial changes associated with GC. In addition, we conducted additional analysis using a random forest classifier to evaluate GC stage-specific microorganisms in liquid biopsy samples (gastric juice, saliva, serum, and urine). The results showed relatively high classification accuracies for serum (87.5%), saliva (80.8%), and urine (79.2%) samples, whereas the classification accuracy for gastric juice samples was lower at 64.2%. These findings further support the potential of specific microorganisms as non-invasive biomarkers for predicting GC stages, particularly highlighting the utility of EVs from liquid biopsy samples such as serum and saliva as diagnostic tools. However, additional data supplementation and improvements in analytical algorithms are necessary to achieve clearer stage differentiation across all sample types.
This study utilized EV-based microbiota analysis to uncover novel microbial dynamics associated with gastric carcinogenesis. Unlike traditional approaches analyzing total microbial DNA from gastric juice or saliva, EV-based analysis focuses on vesicle-encapsulated genetic material, ensuring higher specificity and functional relevance [22,24]. EVs, bacterial-derived structures containing genetic material, proteins, and metabolites, protect microbial information from degradation and facilitate intercellular communication, even in hostile conditions [25,26]. This makes EVs ideal for non-invasive biomarker analysis in liquid biopsies [26]. Serum EVs reflect systemic microbial shifts in advanced disease states, while urine EVs offer a stable medium for profiling localized and systemic changes. By encapsulating active microbial components, EVs highlight functional roles in host-pathogen interactions, adding mechanistic insights to microbiota profiling [24,26]. EV-based methods surpass direct 16S rRNA sequencing by capturing disease-relevant signals with greater precision, particularly from pathogenic microorganisms [22,24,26]. These features underscore the potential of EVs as biomarkers and mediators of gastric carcinogenesis, paving the way for future research and clinical applications [22,26].
Although not well established, we explored the potential roles of these bacteria in gastric pathology through a literature review. Mannosylerythritol lipids secreted by P. antarctica inhibit inflammatory mediators and produce anti-inflammatory effects, which could either contribute to or result from the pathophysiological changes in cancer [27]. Although C. acnes is traditionally linked to skin conditions, its increased abundance in GC raises questions regarding its systemic role, potentially as an agent of inflammation or as a response to the tumor microenvironment [28-30]. Similarly, S. oralis may have implications in GC progression, given its oncogenic potential in the oral cavity and its increased presence in gastric juice with disease progression [31,32]. The systemic increase in P. antarctica, R. insidiosa, and P. yamanorum highlights the potential of these microbes as biomarkers for GC and its precursors, including dysplasia, although their roles in the disease process warrant further exploration [33-36].
Efforts to understand the pathogenesis of GC and to enhance diagnostic methods have greatly benefited from new perspectives and insights gained through extensive research on various biofluids from patients at different stages of the disease [4,37]. Notably, the liquid biopsy approach, which utilizes next-generation sequencing, is a noninvasive alternative that may complement conventional endoscopic examination [38]. Our study extends the well-known association between H. pylori infection and GC by identifying additional bacterial species that could be implicated in the broader dysbiotic shift that accompanies or potentially contributes to cancer development. We highlight the potential of microbial-derived EVs and specific microbial signatures, such as C. acnes, S. oralis, P. antarctica, R. insidiosa, and P. yamanorum, as non-invasive biomarkers for the early detection and monitoring of GC. By analyzing these microbial signatures across various liquid biopsy samples, including gastric juice and extragastric fluids, our study highlights a novel and impactful approach for developing diagnostic tools for GC.
Despite these strengths, some limitations must be acknowledged. Firstly, the control group was relatively small and comprised younger individuals compared to the disease group because efforts to include healthy controls with normal gastric conditions, matched to the age range of the patient groups, were challenging. Although we attempted to mitigate these differences through statistical adjustments for age and sex (Bootstrap, SMOTE, MaAsLin2), smaller and less diverse control groups may have affected the generalizability of our findings. Secondly, while this study demonstrates significant associations between microbial diversity and composition and disease progression in gastric carcinogenesis, it does not establish a direct causal relationship between these factors. The observed differences in microbial diversity and composition may either influence disease progression or result from it, and establishing causality requires further investigation. Experimental models and multi-omics approaches are essential for understanding the role of microbial shifts in gastric carcinogenesis, potentially guiding diagnostic and therapeutic advancements to improve patient outcomes. Thirdly, one critical factor influencing microbial diversity and composition is H. pylori infection, which is well-known to cause dysbiosis and impact gastric disease progression, including cancer [39]. In this study, the low prevalence of H. pylori infection among participants suggests a limited impact on the overall results. To validate our findings, we performed additional analyses by excluding samples from H. pylori-infected individuals. Minor changes were observed, but the primary outcomes remained consistent, suggesting that our findings are not substantially influenced by H. pylori infection status. Lastly, although we identified distinct microbial signatures associated with gastric carcinogenesis, this study did not include functional analyses or an assessment of diagnostic capabilities. Future research is required to explore the functional roles of these microbial signatures in EV-mediated communication pathways and evaluate their diagnostic potential. To strengthen the validity and applicability of the identified biomarkers, future studies should involve larger and more diverse cohorts to establish the robustness and generalizability of the findings.
In conclusion, our study revealed significant changes in the microbial diversity and distinct microbial signatures during gastric carcinogenesis, which were consistently observed in both gastric juice and extragastric liquid biopsy samples. These findings underscore the potential of microbial derived EVs as early diagnostic tools for GC and its precursors. Our study provides a valuable foundation for future studies aimed at utilizing these microbial signatures to enhance early detection and improve diagnostic accuracy. Although the development of non-invasive biomarkers for early GC detection presents challenges, our study provides important insights and highlights the need for continued research to establish reliable and broadly applicable diagnostic tools.
KEY MESSAGE
1. Microbial diversity, particularly alpha and beta diversities, varies across different stages of gastric carcinogenesis, with notable changes in gastric juice and serum.
2. Distinct microbial signatures, including specific bacterial species like C. acnes, S. oralis, and P. antarctica, show unique abundance patterns associated with disease progression.
3. Microbial-derived EVs from liquid biopsy samples, such as gastric juice and extragastric fluids, have the potential to serve as noninvasive biomarkers for the early detection of GC.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement table 1
Supplement table 1 Print
Print





